

Chromatin Immunoprecipitation Protocol & Troubleshooting
Chromatin immunoprecipitation (ChIP) is an immunoprecipitation technique ChIP is based on the use of antibodies to study specific protein-DNA interactions. Antibodies are used to immunoprecipitate the protein of interest and its associated DNA. This powerful method provides valuable information about the interaction between certain proteins and specific DNA sequences, as well as insight into the regulation of gene expression.
Creative Biolabs provides an optimized high-performance solution to meet ChIP. We describe a useful ChIP protocol that includes complete reagents, detailed steps and a troubleshooting guide for optimizing experiments. You can browse the information below to learn more about how to optimize ChIP.
Solutions and Reagents
| Stages | Solutions and Reagents |
| Sample Preparation | Phosphate buffer (PBS), crosslinking agent, termination buffer, lysis buffer |
| Immunoprecipitation | Dilution buffer, antibody solution, magnetic or agarose beads |
| Washing and Elution | Washing buffer, elution buffer, protease |
Chromatin Immunoprecipitation Procedure
ChIP involves a variety of proteomics and molecular biology methods. It involves cross-linking of proteins to DNA, fragmentation, preparation of soluble chromatin and immunoprecipitation with antibodies that recognize the target protein. The following steps describe the details.
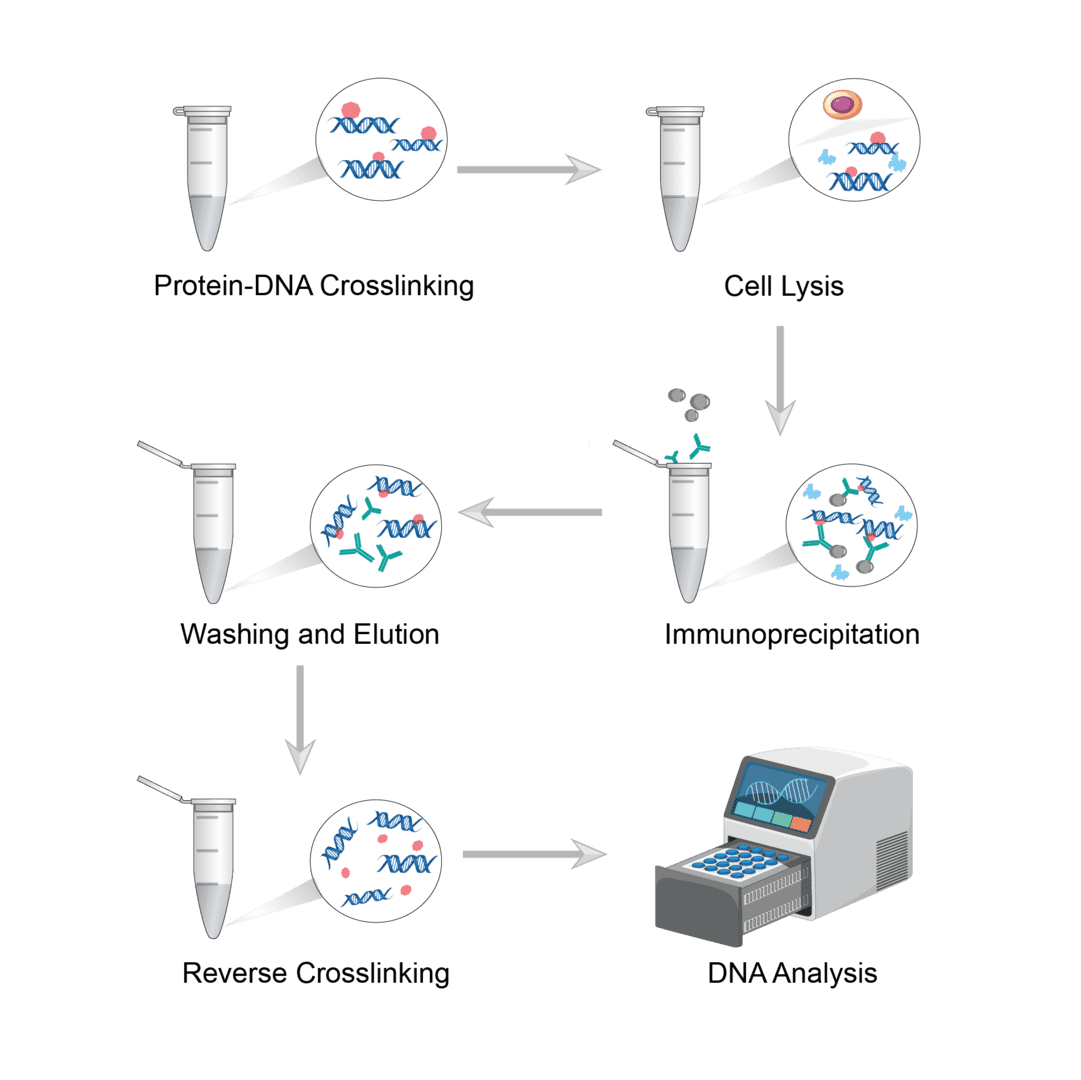
1. Protein-DNA Crosslinking
The use of crosslinkers penetrates into the cell and locks the protein and DNA together. In vivo crosslinking has traditionally been achieved with formaldehyde. Of course, you can also choose other suitable crosslinkers depending on the sample. Dilute the cells and incubate with the crosslinking agent to crosslink the protein-DNA complex. Then add termination buffer to quench the crosslinker. Shake and mix to precipitate the cells, and finally remove the medium.
2. Cell Lysis
Add lysis buffer to resuspend the cell precipitate. Pipette up and down and incubate. Then sonicate the cell suspension and shear the chromatin to a suitable average length. Optimize sonication conditions according to different cell lines. After sonication, centrifuge the sample to precipitate cell debris. Finally, the supernatant is collected as the chromatin preparation to be subsequently analyzed.
3. Immunoprecipitation
Add dilution buffer to dilute the supernatant. Then add the antibody solution to the sample and incubate at room temperature. Then, you can add magnetic or agarose beads to the samples and immunoprecipitate them overnight on a rotating device.
4. Washing and Elution
If using magnetic beads, leave the tube in the magnet to collect the beads. If using agarose beads, collect the beads by centrifugation. Then perform several washes using washing buffer. After washing, then add elution buffer to the bead precipitate and slowly vortex to elute the complexes. Then centrifuge the sample and collect the supernatant.
5. Reverse Crosslinking
The crosslinking between protein and DNA must be reversed prior to analysis of the specific DNA product. You can digest the proteins by thermal incubation or protease. Or use a DNA purification kit to clean up and concentrate the DNA preparation.
6. DNA Analysis
The DNA products obtained are usually measured by real-time quantitative PCR. Alternatively, sequencing can be used for analysis.
Troubleshooting
Below you will find some solutions that can be used to solve common problems observed in ChIP applications. We hope you will find this information useful.
No or low signal
- Not enough cell causes. Too little starting material will produce poor results. We recommend using 25 ug of chromatin per IP and 3-4 million cells. Please use more starting material.
- Excessive crosslinking time causes. Cells may be crosslinked for too long resulting in low signal. Crosslink with formaldehyde for 10-15 minutes and then rinse well with PBS. Excessive crosslinking can reduce epitope availability and thus antibody binding.
- Ineffective cell lysis causes. Cells are not effectively lysed. Insufficient lysis will result in low signal. We recommend lysing cells with a high-quality buffer to improve cell lysis.
- Too small chromatin fragment causes. Excessive sonication can lead to fragment sizes that are too small, which can produce poor results. Do not sonicate chromatin to fragment sizes smaller than 500 bp. Optimize the sonication time to produce fragments between 200-1000 bp.
- Antibody causes. You may not be using the correct antibody. Epitopes may be masked during crosslinking, preventing epitope recognition. We recommend trying polyclonal antibodies or high-quality monoclonal antibodies, which increase the chance of recognition. Also, there may not be enough antibodies in the immunoprecipitation. You can increase the amount of antibody used to enhance the signal. 3-5 µg is usually sufficient, but up to 10 µg may be needed if no signal is observed.
- Wrong affinity bead causes. You may have used the wrong antibody affinity beads. Proteins A and G are bacterial proteins that bind various types of immunoglobulins with different affinities. We recommend using a mixture of protein A and protein G coupled to agarose.
- Strict wash buffer causes. Wash buffers are too stringent. Wash buffers with too high osmotic pressure will reduce antibody binding activity. We recommend using buffers with no more than 500 mM of salt.
High background
- Non-specific binding causes. The first is non-specific binding to protein A or G beads. You can take a pre-clearance step. Incubate the sample with the beads alone to remove the non-specifically bound protein. This is then removed before adding the antibody. Next is an excess of antibody resulting in binding to non-target. And we recommend optimizing the antibody concentration.
- Bead causes. Some protein A or G beads will produce high backgrounds. Use good quality beads to provide the cleanest results with low background in non-specific controls.
- Buffer causes. Contaminated buffers can lead to increased background. Prepare fresh lysis and wash buffer to eliminate this source of error.
ChIP is a powerful method for probing the interaction between proteins and DNA. Understanding and using the correct protocol and addressing common problems is critical to success. For comprehensive or personalized experimental resources, please contact us.
For research use only. Not intended for any clinical use.
Send Inquiry
This site is protected by reCAPTCHA and the Google Privacy Policy and Terms of Service apply.