Since the first therapeutic antibody Muromonab-CD3 was approved in 1986, more than 100 antibodies have been approved for marketing. Based on their properties, antibodies can be divided into four categories: mouse antibodies, chimeric antibodies, humanized antibodies and human antibodies. The first three categories of antibodies are generated by the hybridoma technique. But with the development of phage display, humanized mice, and single B cell technology, more and more fully human antibodies have been approved. Up to now, 36 fully human antibodies have been approved, including recently approved Amivantamab, a bispecific antibody anti EGFR & cMET.
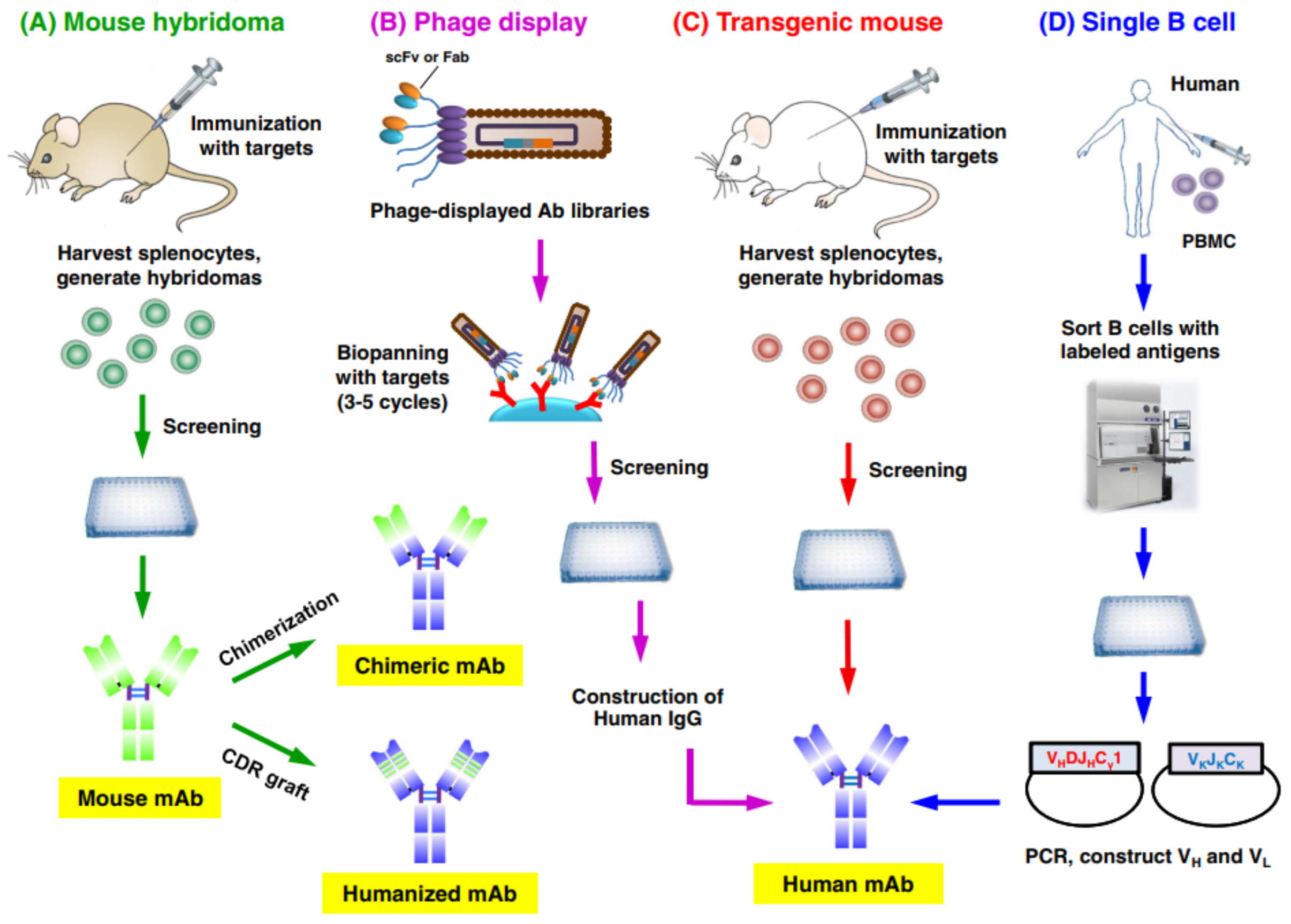
Hybridoma
In 1975, Kohler and Milstein found that hybrid cells generated by fusing mouse myeloma cells with sheep red blood cells-immunized mouse spleen cells not only can produce antibodies but also proliferate indefinitely, thus establishing the hybridoma technology to produce monoclonal antibodies. This discovery won them the Nobel Prize in Medicine and Physiology in 1984.
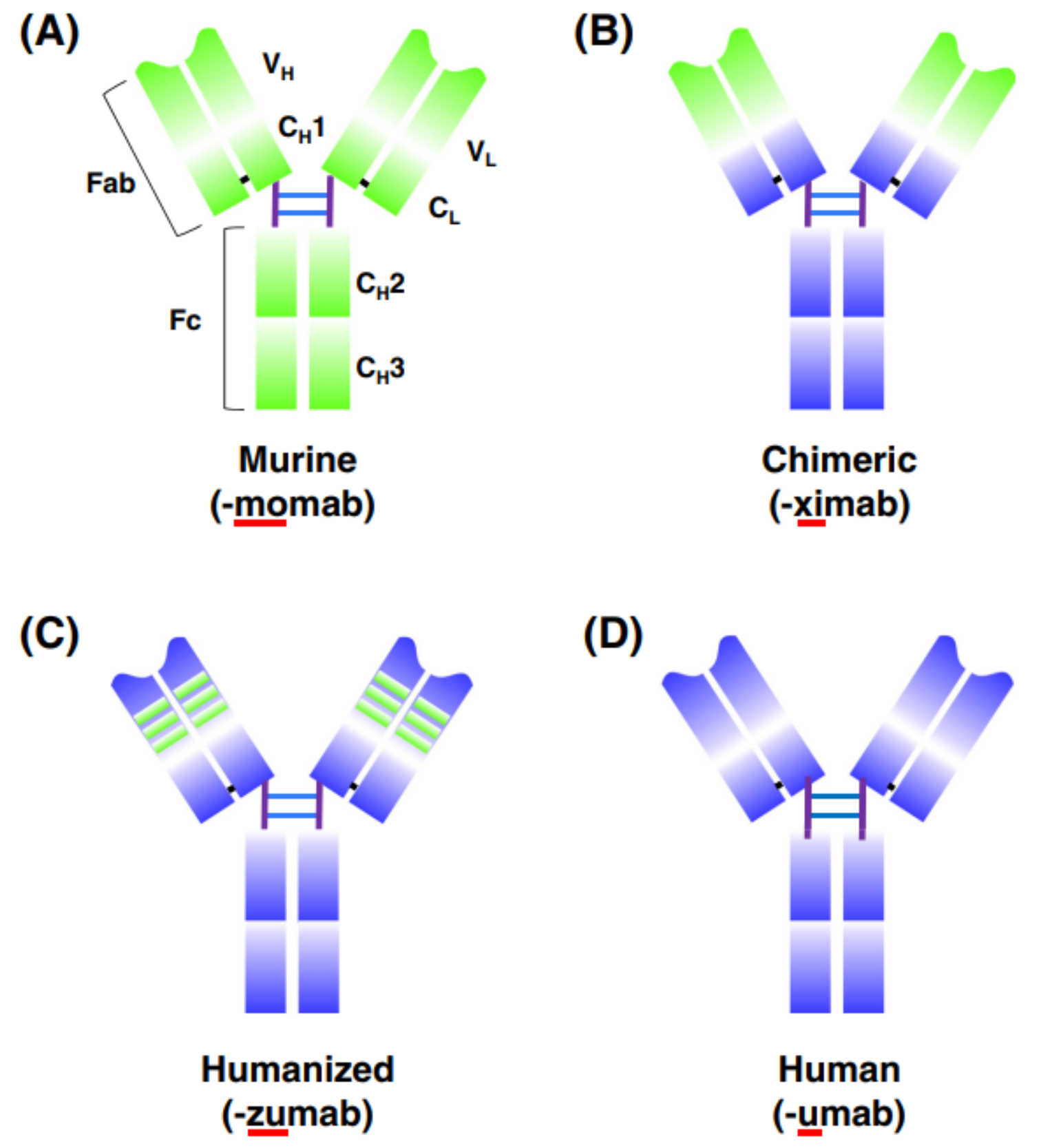
At present, most of the therapeutic antibodies that have been approved for sale are produced by mouse hybridoma cells, because the development of mouse antibodies is more efficient and cost-effective. However, if the mouse antibody is not humanized, it will cause a human anti-mouse antibody (HAMA) response. HAMA not only causes the antibody to be quickly removed but also has potential side effects. Antibody humanization is an efficient way to reduce the HAMA response of mouse monoclonal antibodies, generating human/mouse chimeric antibodies, or humanized antibodies.
The chimeric antibody combines the light chain variable region (VL) and heavy chain variable region (VH) of mouse antibody with the constant region of human antibody through antibody recombination technology. The chimeric antibody not only retains the affinity and high specificity, but also can reduce the mouse origin of the antibody by 70%, greatly reducing the immunogenicity in the human body. The first approved chimeric antibody Abciximab, which targets GPIIb/IIIa, is a Fab fragment approved for marketing in 1994.
The most common practice of humanized antibodies is to transplant the complementarity-determining region (CDR) of mouse antibodies into the framework of human antibodies, which was developed by Gregory P. Winter in 1986. Compared with chimeric antibodies, the degree of human origin of CDR-transplanted antibodies was further improved. Dalizumab (daclizumab) is the first approved antibody to be humanized by CDR transplantation. It binds to IL-2 and is designed to inhibit transplant rejection. In order to reduce the decrease of the affinity of antibodies in the humanization process, the developers of dalizumab chose the antibodies with the most similar structure to mouse antibodies. Humanization is not to simply transplant the CDR region, because in some cases, some amino acids in the non-CDR region of mouse antibodies are essential for antibody binding, and these sites may be involved in the formation of antigenic determinants or directly involved in the binding of antigens. After CDR transplantation, these key sites need to be mutated back to the original amino acid of mouse antibody, which not only maintains the affinity, but also the stability of the antibody.
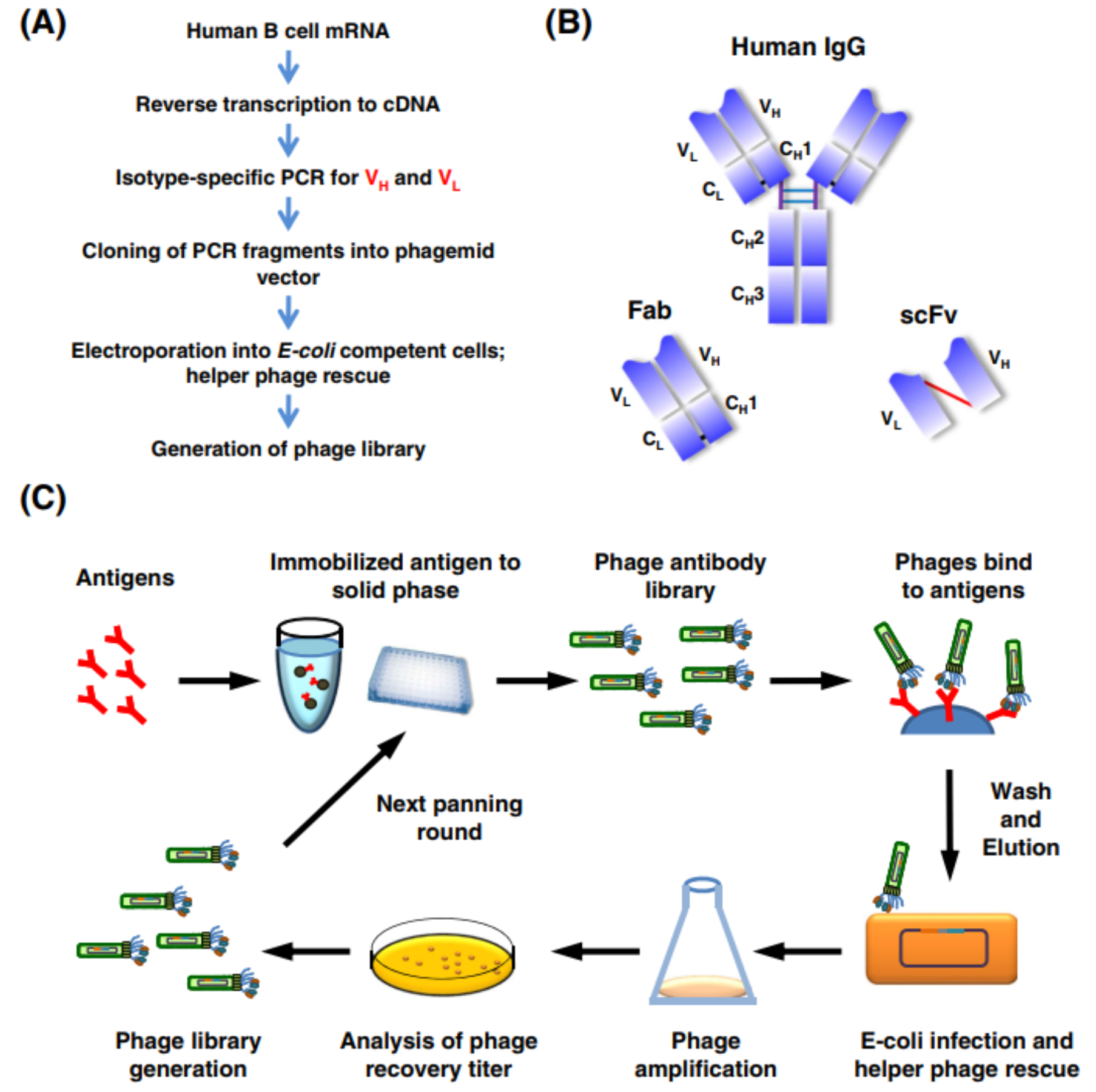
Acquisition of human antibodies by phage display
Phage display technology is the most popular method to screen antibodies in vitro. This technique was developed by George P. Smith in 1985. In phage display, the genes of foreign proteins (e.g. antibodies) are cloned into a vector to be expressed (or displayed) with the structural protein (usually pIII) of M13 phage. Antibodies with high affinity can be screened from display libraries with tens of millions of antibodies.
First, mRNA is obtained from human peripheral blood, and cDNA is obtained by reverse transcription. Then, the VL and VH of all human antibodies are obtained by PCR with a variety of primers. These variable regions are then cloned into vectors with different linker sequences (e.g. for scFv, a flexible linker needs to be added between VH and VL). The resulting vector is transferred into E. coli and the phage library expressing antibody is constructed with the assistance of helper phage.
The antibody genes used to construct the phage library can be obtained from human peripheral blood or immunized animals, or can be comprehensively constructed by random CDR sequences. For the phage library without the former immunization process, the variable region sequence mainly comes from IgM antibodies. The phage library with a former immunization process has a higher probability to screen high-affinity antibodies, because the antibodies go through the stage of in vivo affinity maturation. However, the disadvantage is that different phage libraries need to be constructed for different antibody targets, and the antigens must be able to stimulate the response of the immune system.
Repeated screening can enrich antibodies with high specificity and affinity. Although it may take several weeks, it is still much faster than using the hybridoma technique.
Humanized mouse
Compared with other antibody discovery techniques, humanized mice have many advantages, such as no need for humanization (compared with mouse antibodies), more types of antibodies against the targets, no need for experiments such as affinity maturation, but the threshold is higher because human antibody-related genes need to be introduced into mice.
Alt et al first proposed to use transgenic mice to produce fully human antibodies in 1985; in 1989, Brüggemann and his collaborators first introduced vectors containing two VH genes and different D genes, linked with heavy chain junction region (JH) and μ constant region, into fertilized eggs of mice by microinjection. Only 4% of B lymphocytes in transgenic mice could detect the expression of human μ gene. In 1992, Taylor et al constructed transgenic mice that could express both Vκ-Jκ-Cκ and VH-D-JH-Cμ-Cγ1, but only 10% of the antibodies expressed in these mice were human antibodies. Until 1994, Longberg et al constructed the first human Ig transgenic mouse, HuMab Mouse. They transferred human IgH and IgK genes into mouse IgH and IgK knockout mice. The genomes of human IgH and Ig genes were about 1.29Mb and 1.39Mb, but the genes transferred into mice were smaller than 80Kb, so the diversity of antibodies was much less than that of human. In 1997, Mendez et al transferred yeast artificial chromosomes into mouse embryonic stem cells and hybridized them with IgH and IgK knockout mice to obtain fully-humanized XenoMouse mice, which expressed only human antibodies in vivo.
Although HuMab Mouse and XenoMouse solved the problem of interference of mouse internal antibodies to human antibodies and expand the antibody diversity, because of the lack of mouse Fc, the expression of antibodies in mice, the conversion efficiency of Ig subtypes, and the rate of high-frequency mutation in antibody evolution are relatively low. In order to solve this problem, the researchers connected the Fab region of human antibody with the constant region of mouse antibody, and constructed chimeric humanized mice. Based on this scheme, there are already OmniRat, KyMouse, VelocImmune mouse, H2L2 Mouse and Trianni Mouse.
The first monoclonal antibody developed by humanized mice was approved to market in 2006, and by 2020, as many as 22 monoclonal antibodies produced by humanized mice had been approved for marketing.
Screening of antibodies by single B cell technique
Single B cell technique for antibody screening refers to directly sorting B cells by microfluidics, laser capture microdissection or fluorescence activation sorting, and then amplifying the antibody genes of a single B cell by PCR. Conventional hybridoma or humanized mice need early long-term immunization to screen antibodies. In contrast, single B-cell technology does not need immunization because it separates B cells directly from the human body with good diversity. The natural pairing of antibody heavy chain and light chain is retained.
The preparation of single B cells begins with the identification and isolation of B cells. B cells from peripheral blood, lymph nodes and other tissues are isolated by density gradient centrifugation or flow cytometry. Different separation methods have their unique characteristics. Using density gradient centrifugation results in a mixture of B cells, so the specificity is poor. The B cells isolated by fluorescence antigens such as flow cytometry are B cells that can specifically bind to antigens.
After the isolated B cells are lysed, the antibody genes are amplified by nested or semi-nested RT-PCR with suitable primers. This process requires primers to be universal, sensitive and specific, which can avoid non-specific amplification and amplify complete antibody gene sequences, so the quality of primers directly determines the amplification efficiency of antibody genes. The amplified antibody sequences need to be constructed into a eukaryotic or prokaryotic vector for identification after expression.
Using this technology, Ansuvimab was the first antibody approved for marketing on December 22, 2020, for the treatment of Ebola virus infections in adults and children, developed by Ridgeback Biotherapeutics. In addition to Ansuvimab, there are a number of monoclonal antibodies isolated from human B cells in clinical practice, such as HIV-neutralizing 3BNC117, VRC 01 and PGT121 (isolated from elite patients with HIV), influenza virus-neutralizing MHAA4549A (isolated from people vaccinated with influenza).
Summary
At present, the therapeutic antibody industry is developing rapidly, and more than 100 antibodies, most originated from mice, have been approved for the treatment of tumors, autoimmune diseases, infectious diseases, and so on. With the continuous improvement and maturity of phage display, humanized mice, and single B cell technologies, more and more fully human therapeutic antibodies developed will be approved for marketing. Compared with chimeric antibodies and humanized antibodies, fully human antibodies have a lower risk of immunogenicity, so they are bound to occupy a dominant position in the future.
Reference
1. Lu, Ruei-Min, et al. “Development of therapeutic antibodies for the treatment of diseases.” Journal of biomedical science 27.1 (2020): 1-30.