

DNA Repair
Product List
+ Filter
 Loading...
Loading...- AbPlus™ Anti-APEX1 Magnetic Beads (VS-0724-YC1179) (VS-0724-YC1179)
-
- Target: APEX1
- Target Species: Human
- Application: IP, Protein Purification
 Compare
Compare
- AbPlus™ Anti-Apex1 Magnetic Beads (VS-0724-YC1178) (VS-0724-YC1178)
-
- Target: Apex1
- Target Species: Mouse
- Application: IP, Protein Purification
Compare
- AbPlus™ Anti-XRCC6 Magnetic Beads (VS-0724-YC1116) (VS-0724-YC1116)
-
- Target: XRCC6
- Target Species: Human
- Application: IP, Protein Purification
Compare
- AbPlus™ Anti-MSH2 Magnetic Beads (VS-0724-YC845) (VS-0724-YC845)
-
- Target: MSH2
- Target Species: Human
- Application: IP, Protein Purification
Compare
- AbPlus™ Anti-MBD4 Magnetic Beads (VS-0724-YC156) (VS-0724-YC156)
-
- Target: MBD4
- Target Species: Human
- Application: IP, Protein Purification
Compare
- AbPlus™ Anti-MGMT Magnetic Beads (VS-0724-YC140) (VS-0724-YC140)
-
- Target: MGMT
- Target Species: Human
- Application: IP, Protein Purification
Compare
- AbPlus™ Anti-NBN Magnetic Beads (CBACN-422) (VS-0424-XY198)
-
- Target: NBN
- Target Species: Human, Mouse, Rat
- Application: IP, Protein Purification
Compare
-
- Derivation: Mouse
- Species Reactivity: Human
- Type: Mouse IgG2b, κ
- Application: ELISA, WB, IHC
Compare
-
- Derivation: Mouse
- Species Reactivity: Human
- Type: Mouse IgG2a, κ
- Application: IHC, WB, ELISA
Compare
- Mouse Anti-RNF4 Recombinant Antibody (VS3-FY2863) (VS3-FY2863)
-
- Species Reactivity: Human
- Type: Mouse IgG
- Application: WB
Compare
- Anti-MGMT Recombinant Antibody (VS3-FY2800) (VS3-FY2800)
-
- Type: IgG
- Application: WB, IP, FC
Compare
- Rabbit Anti-CLU Recombinant Antibody (clone R07-4K8) (VS3-XY3320)
-
- Species Reactivity: Human, Mouse
- Type: Rabbit IgG
- Application: WB, IHC-P
Compare
- Recombinant Mouse Anti-XRCC6 Antibody (VS-0923-FY158) (VS-0923-FY158)
-
- Species Reactivity: Human
- Type: Mouse IgG2a
- Application: ELISA, WB
Compare
- Recombinant Mouse Anti-CLU Antibody (VS-0923-FY41) (VS-0923-FY41)
-
- Species Reactivity: Human
- Type: Mouse IgG1
- Application: ELISA, WB
Compare
- Human Anti-CLU Recombinant Antibody (VS-0723-WK113) (VS-0723-WK113)
-
- Derivation: Chimeric (rabbit/human)
- Species Reactivity: Human
- Type: Chimeric (rabbit/human) IgG1
- Application: FC
Compare
- Human Anti-DNA-PKcs Recombinant Antibody (VS-0723-CJ2) (VS-0723-CJ2)
-
- Derivation: Humanized
- Species Reactivity: Human
- Type: Human IgG
- Application: ELISA
Compare
- Recombinant Rabbit Anti-NBN Antibody (clone R04-7C7) (VS3-FY1003)
-
- Species Reactivity: Human
- Type: Rabbit IgG
- Application: WB, ICC, IF, IP
Compare
-
- Derivation: Mouse
- Species Reactivity: Human, Mouse
- Type: Mouse IgG2a
- Application: WB, ICC, IHC, FC
Compare
-
- Derivation: Mouse
- Species Reactivity: Human, Mouse
- Type: Mouse IgG2b
- Application: WB, ICC, IHC, FC
Compare
-
- Derivation: Mouse
- Species Reactivity: Human, Mouse
- Type: Mouse IgG1
- Application: WB, IHC, FC
Compare
-
- Derivation: Mouse
- Species Reactivity: Human, Mouse, Rat
- Type: Mouse IgG1
- Application: WB, IP
Compare
-
- Derivation: Mouse
- Species Reactivity: Human, Mouse, Rat, Monkey, Hamster
- Type: Mouse IgG1
- Application: WB, ICC
Compare
-
- Derivation: Mouse
- Species Reactivity: Human
- Type: Mouse IgG1
- Application: WB, IHC, ICC, FC, ELISA
Compare
-
- Derivation: Mouse
- Species Reactivity: Human, Monkey
- Type: Mouse IgG1
- Application: WB, ICC, IP
Compare
-
- Derivation: Mouse
- Species Reactivity: Human
- Type: Mouse IgG1
- Application: WB, IHC, ICC, FC, ELISA
Compare
-
- Derivation: Mouse
- Species Reactivity: Human, Monkey, Rat
- Type: Mouse IgG1
- Application: WB, ELISA
Compare
- Mouse Anti-MBD4 Recombinant Antibody (VS3-CJ106) (VS3-CJ106)
-
- Species Reactivity: Human
- Type: Mouse IgG
- Application: WB, ELISA
Compare
- Rabbit Anti-NBN Recombinant Antibody (VS3-CJ165) (VS3-CJ165)
-
- Species Reactivity: Human
- Type: Rabbit IgG
- Application: WB, ICC, IF, IHC, IP
Compare
- Rabbit Anti-NBN Recombinant Antibody (VS3-CJ166) (VS3-CJ166)
-
- Species Reactivity: Human, Mouse, Rat
- Type: Rabbit IgG
- Application: WB, ICC, IF, IP, FC, IHC, CHIP
Compare
- Mouse Anti-MGMT Recombinant Antibody (VS3-CJ702) (VS3-CJ702)
-
- Species Reactivity: Human
- Type: Mouse IgG1
- Application: WB, IF, IHC-P
Compare
- Mouse Anti-MSH2 Recombinant Antibody (VS3-CJ961) (VS3-CJ961)
-
- Species Reactivity: Human
- Type: Mouse IgG
- Application: WB, IHC
Compare
- Mouse Anti-MGMT Recombinant Antibody (clone 4A2) (VS3-XY1071)
-
- Species Reactivity: Human
- Type: Mouse IgG1
- Application: ELISA, WB
Compare
- Mouse Anti-MGMT Recombinant Antibody (clone 5A2G8) (VS3-XY1072)
-
- Species Reactivity: Human
- Type: Mouse IgG1
- Application: ELISA, WB
Compare
- Mouse Anti-NBN Recombinant Antibody (clone 7E4A2) (VS3-XY1135)
-
- Species Reactivity: Human
- Type: Mouse IgG2a
- Application: ELISA, WB, IHC, FC, ICC
Compare
- Mouse Anti-NBN Recombinant Antibody (clone 7E4C2) (VS3-XY1136)
-
- Species Reactivity: Human
- Type: Mouse IgG2a
- Application: ELISA, WB, IHC, FC, ICC
Compare
- Mouse Anti-OGG1 Recombinant Antibody (clone 1G4) (VS3-XY1196)
-
- Species Reactivity: Human
- Type: Mouse IgG1
- Application: WB, IP, IF
Compare
- Mouse Anti-XRCC6 Recombinant Antibody (clone 2F7F5) (VS3-XY1608)
-
- Species Reactivity: Human
- Type: Mouse IgG1
- Application: ELISA, WB, IHC, FC, ICC
Compare
- Mouse Anti-XRCC6 Recombinant Antibody (clone 6C8G7) (VS3-XY1609)
-
- Species Reactivity: Human
- Type: Mouse IgG1
- Application: ELISA, WB, FC, ICC
Compare
- Mouse Anti-XRCC6 Recombinant Antibody (clone 8D5) (VS3-XY1610)
-
- Species Reactivity: Human
- Type: Mouse IgG1
- Application: ELISA, WB, IHC, FC, ICC
Compare
- Mouse Anti-BRCA1 Recombinant Antibody (VS3-WK1325) (VS3-WK1325)
-
- Derivation: Mouse
- Species Reactivity: Human
- Type: Mouse IgG
- Application: ELISA, WB
Compare
View More Products
Our customer service representatives are available 24 hours a day, from Monday to Sunday. Contact Us
Can't find the products you're looking for? Try to filter in the left sidebar.Filter By Tag
Representative Targets in DNA Repair Full List of Targets in DNA Repair Tested Data-Supported Products for Targeting DNA Repairt
DNA repair is an essential cellular mechanism that safeguards the genetic material from damage caused by environmental factors, such as ultraviolet radiation and chemical mutagens, as well as endogenous threats, including reactive oxygen species and replication errors. This complex process is critical for maintaining genomic stability, preventing mutations that could lead to cellular dysfunction, aging, and diseases such as cancer. DNA repair mechanisms are diverse, including direct reversal, base excision repair, nucleotide excision repair, mismatch repair, and homologous recombination, each tailored to address specific types of DNA damage. These pathways operate through a highly coordinated effort of detection, signaling, and repair, involving a multitude of enzymes and proteins that recognize and correct damaged DNA. The role of DNA repair extends beyond merely fixing genetic lesions; it is integral in cell cycle regulation, where it acts at checkpoints to halt progression until damage is resolved, thereby ensuring accurate DNA replication and division. Furthermore, DNA repair mechanisms are pivotal in the immune system, contributing to the generation of antibody diversity through somatic hypermutation and class switch recombination. The effectiveness of DNA repair is a balancing act between repairing harmful lesions and avoiding excessive repair that could lead to deleterious mutations, highlighting its intricacy and significance in cellular homeostasis and the prevention of disease.
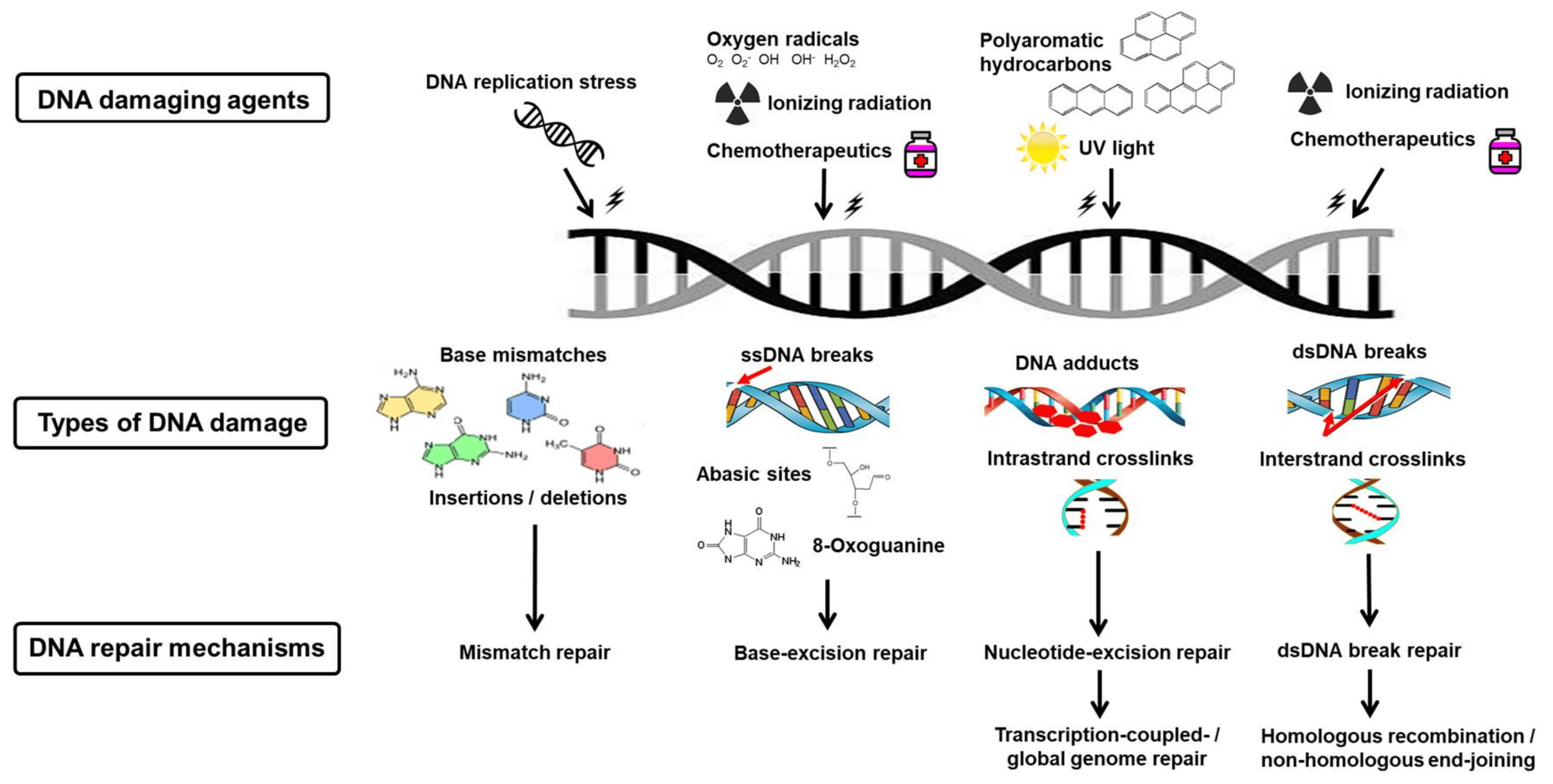 Figure 1 DNA damage and repair mechanisms. (Helena, 2018)
Figure 1 DNA damage and repair mechanisms. (Helena, 2018)
Representative Targets in DNA Repair
MCM2
MCM2 (Minichromosome Maintenance Complex Component 2) is an essential protein that plays a pivotal role in the initiation and regulation of DNA replication, a critical process for genetic inheritance and cell division. It is a part of the MCM complex, which consists of six highly conserved proteins (MCM2-7) forming a hexameric ring structure that functions as a helicase during DNA replication. This helicase activity is crucial for unwinding the double-stranded DNA, thereby providing the single-stranded DNA templates necessary for DNA polymerase to synthesize new DNA strands. The activation and regulation of the MCM complex are tightly controlled within the cell cycle, primarily at the G1 to S phase transition, ensuring that DNA replication occurs once and only once per cell cycle. This is vital for maintaining genomic integrity and preventing DNA replication errors, which could lead to genomic instability and contribute to disease progression, including cancer. MCM2, in particular, has been identified not only as a key player in DNA replication initiation but also in the replication stress response, where it participates in mechanisms designed to protect replicating DNA under conditions of stress. The expression levels of MCM2 have been found to be elevated in various types of cancer, serving as a potential biomarker for cancer diagnosis and prognosis.
Recommended Mouse Anti-MCM2 mAb (CAT#: MOB-0101F)
 Figure 2 Mouse Anti-MCM2 Recombinant Antibody (MOB-0101F) in IHC. Immunohistochemical analysis of paraffin-embedded colon cancer. 1. The antibody is diluted 1:200 (overnight at 4°C). 2. Use pH8.0 TRIS-EDTA for antigen retrieval. 3. Dilute the secondary antibody at 1:200 (room temperature, 30min).
Figure 2 Mouse Anti-MCM2 Recombinant Antibody (MOB-0101F) in IHC. Immunohistochemical analysis of paraffin-embedded colon cancer. 1. The antibody is diluted 1:200 (overnight at 4°C). 2. Use pH8.0 TRIS-EDTA for antigen retrieval. 3. Dilute the secondary antibody at 1:200 (room temperature, 30min).
Recommended Mouse Anti-MCM2 mAb (CAT#: MOB-0102F)
 Figure 3 Mouse Anti-MCM2 Recombinant Antibody (MOB-0102) in IHC. Immunohistochemical analysis of paraffin-embedded colon cancer. 1. The antibody is diluted 1:200 (overnight at 4°C). 2. Use pH8.0 TRIS-EDTA for antigen retrieval. 3. Dilute the secondary antibody at 1:200 (room temperature, 30min).
Figure 3 Mouse Anti-MCM2 Recombinant Antibody (MOB-0102) in IHC. Immunohistochemical analysis of paraffin-embedded colon cancer. 1. The antibody is diluted 1:200 (overnight at 4°C). 2. Use pH8.0 TRIS-EDTA for antigen retrieval. 3. Dilute the secondary antibody at 1:200 (room temperature, 30min).
Recommended Mouse Anti-MCM2 mAb (CAT#: VS3-FY907)
 Figure 4 Recombinant Rabbit Anti-MCM2 (phospho Ser108) Antibody (clone R01-7G9) in ICC. Immunocytochemical analysis of MCM2 (Phospho-Ser108) (green) in Hela using MCM2 (Phospho-Ser108) antibody and DAPI (blue).
Figure 4 Recombinant Rabbit Anti-MCM2 (phospho Ser108) Antibody (clone R01-7G9) in ICC. Immunocytochemical analysis of MCM2 (Phospho-Ser108) (green) in Hela using MCM2 (Phospho-Ser108) antibody and DAPI (blue).
MSH2
MSH2 (MutS Homolog 2) is a crucial component of the DNA mismatch repair (MMR) system, a highly conserved mechanism essential for maintaining genomic stability by correcting base-pair mismatches and insertion-deletion loops that occur during DNA replication and recombination. As part of the MMR pathway, MSH2 forms heterodimers with other MMR proteins, most commonly MSH6 (to form MutSα) or MSH3 (to form MutSβ), and these complexes play a pivotal role in the initial recognition and binding to mismatches. Following mismatch recognition, the MSH2-containing complexes recruit and interact with other MMR proteins, such as MLH1, to initiate the repair process. This involves the removal of the mismatched segment of DNA followed by resynthesis using the correct template strand, thereby preventing the propagation of replication errors that could lead to mutations. The importance of MSH2 and the MMR system extends beyond DNA repair, as they are also involved in the cellular response to DNA damage, contributing to apoptosis and cell cycle arrest in the presence of irreparable DNA damage. Mutations or deficiencies in MSH2 and other MMR proteins are strongly associated with hereditary nonpolyposis colorectal cancer (HNPCC), also known as Lynch syndrome, which predisposes individuals to a high risk of developing colorectal cancer and other types of cancer at an early age. The identification of MSH2 mutations offers valuable information for cancer risk assessment and the management of affected individuals and their families.
Recommended Mouse Anti-MSH2 mAb (CAT#: ZG-0289C)
 Figure 5 Mouse Anti-MSH2 Antibody (ZG-0289C) in IHC. Immunohistochemistry analysis of paraffin-embedded human breast cancer (left) and lung cancer (right) tissues, showing nuclear localization with DAB staining using MSH2 monoclonal antibody.
Figure 5 Mouse Anti-MSH2 Antibody (ZG-0289C) in IHC. Immunohistochemistry analysis of paraffin-embedded human breast cancer (left) and lung cancer (right) tissues, showing nuclear localization with DAB staining using MSH2 monoclonal antibody.
PRKDC
PRKDC (Protein Kinase, DNA-Activated, Catalytic Subunit), also known as DNA-PKcs (DNA-Dependent Protein Kinase, Catalytic Subunit), is a key enzyme involved in the repair of DNA double-strand breaks (DSBs), one of the most lethal forms of DNA damage. As an integral component of the non-homologous end joining (NHEJ) pathway, PRKDC plays a critical role in maintaining genomic integrity by facilitating the direct re-ligation of DSBs, which can result from ionizing radiation, reactive oxygen species, and various chemical exposures, as well as through normal cellular processes such as V(D)J recombination during the development of the immune system. The protein functions by forming a complex with the KU70/KU80 heterodimer, which initially binds to the ends of DSBs. This binding allows PRKDC to localize to the DNA damage site, where it undergoes autophosphorylation and becomes fully activated. Upon activation, PRKDC phosphorylates a range of downstream proteins involved in the DNA damage response, including itself and factors essential for DNA repair, cell cycle regulation, and apoptosis. This activity not only promotes the repair of DSBs but also modulates the cellular response to DNA damage, including cell cycle arrest and the activation of transcription factors that induce the expression of genes involved in DNA repair and cell survival. Dysregulation or mutations in PRKDC have been implicated in various cancers, as defects in DSB repair can lead to genomic instability and the accumulation of mutations that drive carcinogenesis.
Recommended Mouse Anti-PRKDC mAb (CAT#: ZG-0480F)
 Figure 6 Mouse Anti-PRKDC Recombinant Antibody (ZG-0480F) in IF. Immunofluorescence analysis of Hela cells using DNA-PKCS monoclonal antibody (green).
Figure 6 Mouse Anti-PRKDC Recombinant Antibody (ZG-0480F) in IF. Immunofluorescence analysis of Hela cells using DNA-PKCS monoclonal antibody (green).
Recommended Mouse Anti-PRKDC mAb (CAT#: ZG-0481F)
 Figure 7 Mouse Anti-PRKDC Recombinant Antibody (ZG-0481F) in ICC. Immunocytochemical staining of Hela with DNA-PKcs mouse mAb (1:200).
Figure 7 Mouse Anti-PRKDC Recombinant Antibody (ZG-0481F) in ICC. Immunocytochemical staining of Hela with DNA-PKcs mouse mAb (1:200).
Recommended Mouse Anti-PRKDC mAb (CAT#: ZG-0482F)
 Figure 8 Mouse Anti-PRKDC Recombinant Antibody (ZG-0482) in ICC. Immunocytochemical staining of Hela with DNA-PKcs mouse mAb (1:200).
Figure 8 Mouse Anti-PRKDC Recombinant Antibody (ZG-0482) in ICC. Immunocytochemical staining of Hela with DNA-PKcs mouse mAb (1:200).
Full List of Targets in DNA Repair
| Biomarker | Alternative Names | Gene ID | UniProt ID | Roles |
| APEX1 | Apurinic/Apyrimidinic Endodeoxyribonuclease 1; APEX Nuclease (Multifunctional DNA Repair Enzyme) 1; Apurinic-Apyrimidinic Endonuclease 1; Redox Factor-1; EC 4.2.99.18; APEX; APE1; APEN; HAP1; REF1; APE; APX; APEX Nuclease (Multifunctional DNA Repair Enzyme) | 328 | P27695 | Apurinic/apyrimidinic (AP) sites occur frequently in DNA molecules by spontaneous hydrolysis, by DNA damaging agents or by DNA glycosylases that remove specific abnormal bases. AP sites are pre-mutagenic lesions that can prevent normal DNA replication so the cell contains systems to identify and repair such sites. Class II AP endonucleases cleave the phosphodiester backbone 5' to the AP site. This gene encodes the major AP endonuclease in human cells. Splice variants have been found for this gene; all encode the same protein. |
| BABAM1 | NBA1; HSPC142; MERIT40; C19orf62 | 29086 | Q9NWV8 | |
| BRCA1 | IRIS; PSCP; BRCAI; BRCC1; FANCS; PNCA4; RNF53; BROVCA1; PPP1R53 | 672 | P38398 | This gene encodes a 190 kD nuclear phosphoprotein that plays a role in maintaining genomic stability, and it also acts as a tumor suppressor. The BRCA1 gene contains 22 exons spanning about 110 kb of DNA. The encoded protein combines with other tumor suppressors, DNA damage sensors, and signal transducers to form a large multi-subunit protein complex known as the BRCA1-associated genome surveillance complex (BASC). This gene product associates with RNA polymerase II, and through the C-terminal domain, also interacts with histone deacetylase complexes. This protein thus plays a role in transcription, DNA repair of double-stranded breaks, and recombination. Mutations in this gene are responsible for approximately 40% of inherited breast cancers and more than 80% of inherited breast and ovarian cancers. Alternative splicing plays a role in modulating the subcellular localization and physiological function of this gene. Many alternatively spliced transcript variants, some of which are disease-associated mutations, have been described for this gene, but the full-length natures of only some of these variants has been described. A related pseudogene, which is also located on chromosome 17, has been identified. |
| CLU | Clusterin; Testosterone-Repressed Prostate Message 2; Apolipoprotein J; Complement-Associated Protein SP-40,40; Complement Cytolysis Inhibitor; Complement Lysis Inhibitor; Sulfated Glycoprotein 2; Ku70-Binding Protein 1; NA1/NA2; TRPM-2; APO-J; APOJ; KUB1 | 1191 | P10909 | The protein encoded by this gene is a secreted chaperone that can under some stress conditions also be found in the cell cytosol. It has been suggested to be involved in several basic biological events such as cell death, tumor progression, and neurodegenerative disorders. Alternate splicing results in both coding and non-coding variants.[provided by RefSeq, May 2011] |
| DDB2 | Damage Specific DNA Binding Protein 2; UV-Damaged DNA-Binding Protein 2; DDB P48 Subunit; Xeroderma Pigmentosum Group E Protein; UV-DDB 2; DDBB; Damage-Specific DNA Binding Protein 2 (48kD) | 1643 | Q92466 | This gene encodes a protein that is necessary for the repair of ultraviolet light-damaged DNA. This protein is the smaller subunit of a heterodimeric protein complex that participates in nucleotide excision repair, and this complex mediates the ubiquitylation of histones H3 and H4, which facilitates the cellular response to DNA damage. This subunit appears to be required for DNA binding. Mutations in this gene cause xeroderma pigmentosum complementation group E, a recessive disease that is characterized by an increased sensitivity to UV light and a high predisposition for skin cancer development, in some cases accompanied by neurological abnormalities. Two transcript variants encoding different isoforms have been found for this gene. |
| DNA-PKcs | DNA-dependent protein kinase catalytic subunit | Overexpression of DNA-dependent protein kinase catalytic subunit (DNA-PKcs) is commonly occurred in cancers and causes radioresistance and poor prognosis. | ||
| DNTT | DNA Nucleotidylexotransferase; Terminal Deoxynucleotidyltransferase; Terminal Addition Enzyme; Terminal Transferase; EC 2.7.7.31; TDT; | 1791 | P04053 | This gene is a member of the DNA polymerase type-X family and encodes a template-independent DNA polymerase that catalyzes the addition of deoxynucleotides to the 3'-hydroxyl terminus of oligonucleotide primers. In vivo, the encoded protein is expressed in a restricted population of normal and malignant pre-B and pre-T lymphocytes during early differentiation, where it generates antigen receptor diversity by synthesizing non-germ line elements (N-regions) at the junctions of rearranged Ig heavy chain and T cell receptor gene segments. Alternatively spliced transcript variants encoding different isoforms of this gene have been described. |
| ERCC1 | ERCC Excision Repair 1, Endonuclease Non-Catalytic Subunit; Excision Repair Cross-Complementing Rodent Repair Deficiency, Complementation Group 1 (Includes Overlapping Antisense Sequence); Excision Repair Cross-Complementation Group 1; DNA Excision Repair Protein ERCC-1; COFS4; RAD10; UV20; | 2067 | P07992 | The product of this gene functions in the nucleotide excision repair pathway, and is required for the repair of DNA lesions such as those induced by UV light or formed by electrophilic compounds including cisplatin. The encoded protein forms a heterodimer with the XPF endonuclease (also known as ERCC4), and the heterodimeric endonuclease catalyzes the 5' incision in the process of excising the DNA lesion. The heterodimeric endonuclease is also involved in recombinational DNA repair and in the repair of inter-strand crosslinks. Mutations in this gene result in cerebrooculofacioskeletal syndrome, and polymorphisms that alter expression of this gene may play a role in carcinogenesis. Multiple transcript variants encoding different isoforms have been found for this gene. The last exon of this gene overlaps with the CD3e molecule, epsilon associated protein gene on the opposite strand. |
| ERCC3 | ERCC3; RAD25; Excision Repair Cross-Complementing Rodent Repair Deficiency, Complementation Group 3; XPB; GTF2H; BTF2; TFIIH; BTF2 P89; XPBC; DNA Excision Repair Protein ERCC-3; DNA Repair Protein Complementing XP-B Cells; TFIIH 89 KDa Subunit; Xeroderma | 2071 | P19447 | This gene encodes an ATP-dependent DNA helicase that functions in nucleotide excision repair. The encoded protein is a subunit of basal transcription factor 2 (TFIIH) and, therefore, also functions in class II transcription. Mutations in this gene are associated with Xeroderma pigmentosum B, Cockayne's syndrome, and trichothiodystrophy. Alternative splicing results in multiple transcript variants. |
| ERCC4 | ERCC4; excision repair cross-complementing rodent repair deficiency, complementation group 4; XPF; DNA repair endonuclease XPF; RAD1; xeroderma pigmentosum; complementation group F; DNA excision repair protein ERCC 4; DNA excision repair protein ERCC-4; DNA excision repair protein ERCC4; DNA repair endonuclease XPF; DNA repair protein complementing XP F cells; DNA repair protein complementing XP-F cells; ERCC 11; ERCC 4; ERCC11; ERCC4; Excision repair complementing defective in Chinese hamster; Excision repair cross complementing rodent repair deficiency complementation group 4; RAD 1; RAD1; Xeroderma pigmentosum complementation group F; Xeroderma pigmentosum group F complementing protein; Xeroderma pigmentosum group F-complementing protein; XPF_HUMAN; OTTHUMP00000045868; DNA excision repair protein ERCC-4; DNA repair protein complementing XP-F cells; xeroderma pigmentosum, complementation group F; xeroderma pigmentosum group F-complementing protein; excision-repair, complementing; ERCC11; | 2072 | Q92889 | The protein encoded by this gene forms a complex with ERCC1 and is involved in the 5 incision made during nucleotide excision repair. This complex is a structure specific DNA repair endonuclease that interacts with EME1. Defects in this gene are a cause of xeroderma pigmentosum complementation group F (XP-F), or xeroderma pigmentosum VI (XP6).[provided by RefSeq, Mar 2009] |
| FEN1 | FEN-1; MF1; RAD2 | 2237 | P39748 | The protein encoded by this gene removes 5' overhanging flaps in DNA repair and processes the 5' ends of Okazaki fragments in lagging strand DNA synthesis. Direct physical interaction between this protein and AP endonuclease 1 during long-patch base excision repair provides coordinated loading of the proteins onto the substrate, thus passing the substrate from one enzyme to another. The protein is a member of the XPG/RAD2 endonuclease family and is one of ten proteins essential for cell-free DNA replication. DNA secondary structure can inhibit flap processing at certain trinucleotide repeats in a length-dependent manner by concealing the 5' end of the flap that is necessary for both binding and cleavage by the protein encoded by this gene. Therefore, secondary structure can deter the protective function of this protein, leading to site-specific trinucleotide expansions. [provided by RefSeq, Jul 2008] |
| MBD4 | MED1; TPDS2 | 8930 | O95243 | DNA methylation is the major modification of eukaryotic genomes and plays an essential role in mammalian development. Human proteins MECP2, MBD1, MBD2, MBD3, and MBD4 comprise a family of nuclear proteins related by the presence in each of a methyl-CpG binding domain (MBD). MBD4 may function to mediate the biological consequences of the methylation signal. In addition, MBD4 has protein sequence similarity to bacterial DNA repair enzymes and thus may have some function in DNA repair. |
| MCM2 | Minichromosome Maintenance Complex Component 2; Minichromosome Maintenance Protein 2 Homolog; Nuclear Protein BM28; Mitotin; CCNL1; CDCL1; BM28; MCM2 Minichromosome Maintenance Deficient 2, Mitotin (S. Cerevisiae); Minichromosome Maintenance Deficient (S. Cerevisiae) 2 (Mitotin); Minichromosome Maintenance Deficient 2 (Mitotin) | 4171 | P49736 | The protein encoded by this gene is one of the highly conserved mini-chromosome maintenance proteins (MCM) that are involved in the initiation of eukaryotic genome replication. The hexameric protein complex formed by MCM proteins is a key component of the pre-replication complex (pre_RC) and may be involved in the formation of replication forks and in the recruitment of other DNA replication related proteins. This protein forms a complex with MCM4, 6, and 7, and has been shown to regulate the helicase activity of the complex. This protein is phosphorylated, and thus regulated by, protein kinases CDC2 and CDC7. Multiple alternatively spliced transcript variants have been found, but the full-length nature of some variants has not been defined. [provided by RefSeq, Oct 2012] |
| MED1 | PBP; CRSP1; RB18A; TRIP2; PPARBP; CRSP200; DRIP205; DRIP230; PPARGBP; TRAP220 | 5469 | Q15648 | MED1 work with coactivators to direct transcriptional initiation by the RNA polymerase II apparatus. The protein encoded by this gene is a subunit of the CRSP (cofactor required for SP1 activation) complex, which, along with TFIID, is required for efficient activation by SP1. It also regulates p53 dependent apoptosis and it is essential for adipogenesis. This protein is known to have the ability to self-oligomerize. |
| MGMT | O-6-Methylguanine-DNA Methyltransferase; Methylated-DNA--Protein-Cysteine Methyltransferase; -O-Methylguanine-DNA Methyltransferase; O-6-Methylguanine-DNA-Alkyltransferase; EC 2.1.1.63; O6-Methylguanine-DNA Methyltransferase; Methylguanine-DNA Methyltransferase | 4255 | P16455 | Alkylating agents are potent carcinogens that can result in cell death, mutation and cancer. The protein encoded by this gene is a DNA repair protein that is involved in cellular defense against mutagenesis and toxicity from alkylating agents. The protein catalyzes transfer of methyl groups from O(6)-alkylguanine and other methylated moieties of the DNA to its own molecule, which repairs the toxic lesions. Methylation of the genes promoter has been associated with several cancer types, including colorectal cancer, lung cancer, lymphoma and glioblastoma. [provided by RefSeq, Sep 2015] |
| MSH2 | MutS Homolog 2; HMSH2; MutS (E. Coli) Homolog 2 (Colon Cancer, Nonpolyposis Type 1); MutS Homolog 2, Colon Cancer, Nonpolyposis Type 1 (E. Coli); MutS Homolog 2, Colon Cancer, Nonpolyposis Type 1; DNA Mismatch Repair Protein Msh2; MutS Protein Homolog 2 | 4436 | P43246 | This locus is frequently mutated in hereditary nonpolyposis colon cancer (HNPCC). When cloned, it was discovered to be a human homolog of the E. Coli mismatch repair gene mutS, consistent with the characteristic alterations in microsatellite sequences (RER+ phenotype) found in HNPCC. Two transcript variants encoding different isoforms have been found for this gene. [provided by RefSeq, Apr 2012] |
| NBN | Nibrin; Nijmegen Breakage Syndrome 1 (Nibrin); Cell Cycle Regulatory Protein P95; NBS1; NBS; P95 | 4683 | O60934 | Mutations in this gene are associated with Nijmegen breakage syndrome, an autosomal recessive chromosomal instability syndrome characterized by microcephaly, growth retardation, immunodeficiency, and cancer predisposition. The encoded protein is a member of the MRE11/RAD50 double-strand break repair complex which consists of 5 proteins. This gene product is thought to be involved in DNA double-strand break repair and DNA damage-induced checkpoint activation. [provided by RefSeq, Jul 2008] |
| NTHL1 | Nth Like DNA Glycosylase 1; Bifunctional DNA N-Glycosylase/DNA-(Apurinic Or Apyrimidinic Site) Lyase; DNA Glycosylase/AP Lyase; OCTS3; HNTH1; NTH1; Bifunctional DNA N-Glycoslyase/DNA-(Apurinic Or Apyrimidinic Site) Lyase; Nth Endonuclease III-Like 1 (E. Coli) | 4913 | P78549 | The protein encoded by this gene is a DNA N-glycosylase of the endonuclease III family. Like a similar protein in E. Coli, the encoded protein has DNA glycosylase activity on DNA substrates containing oxidized pyrimidine residues and has apurinic/apyrimidinic lyase activity. [provided by RefSeq, Oct 2008] |
| OGG1 | 8-Oxoguanine DNA Glycosylase; 8-Hydroxyguanine DNA Glycosylase; MUTM; OGH1; DNA-Apurinic Or Apyrimidinic Site Lyase; N-Glycosylase/DNA Lyase | 4968 | O15527 | This gene encodes the enzyme responsible for the excision of 8-oxoguanine, a mutagenic base byproduct which occurs as a result of exposure to reactive oxygen. The action of this enzyme includes lyase activity for chain cleavage. Alternative splicing of the C-terminal region of this gene classifies splice variants into two major groups, type 1 and type 2, depending on the last exon of the sequence. Type 1 alternative splice variants end with exon 7 and type 2 end with exon 8. All variants share the N-terminal region in common, which contains a mitochondrial targeting signal that is essential for mitochondrial localization. Many alternative splice variants for this gene have been described, but the full-length nature for every variant has not been determined. |
| PRKDC | HYRC; p350; DNAPK; DNPK1; HYRC1; IMD26; XRCC7; DNAPKc; DNA-PKC; DNA-PKcs | 5591 | P78527 | This gene encodes the catalytic subunit of the DNA-dependent protein kinase (DNA-PK). It functions with the Ku70/Ku80 heterodimer protein in DNA double strand break repair and recombination. The protein encoded is a member of the PI3/PI4-kinase family. |
| RAD50 | RAD50 Double Strand Break Repair Protein; RAD50 Homolog, Double Strand Break Repair Protein; HRad50; RAD50 (S. Cerevisiae) Homolog; RAD50 Homolog (S. Cerevisiae); DNA Repair Protein RAD50; EC 3.6.3.27 | 10111 | Q92878 | The protein encoded by this gene is highly similar to Saccharomyces cerevisiae Rad50, a protein involved in DNA double-strand break repair. This protein forms a complex with MRE11 and NBS1. The protein complex binds to DNA and displays numerous enzymatic activities that are required for nonhomologous joining of DNA ends. This protein, cooperating with its partners, is important for DNA double-strand break repair, cell cycle checkpoint activation, telomere maintenance, and meiotic recombination. Knockout studies of the mouse homolog suggest this gene is essential for cell growth and viability. Mutations in this gene are the cause of Nijmegen breakage syndrome-like disorder.[provided by RefSeq, Apr 2010] |
| SLC19A1 | CHMD; FOLT; IFC1; REFC; RFC1 | 6573 | P41440 | The membrane protein encoded by this gene is a transporter of folate and is involved in the regulation of intracellular concentrations of folate. Three transcript variants encoding different isoforms have been found for this gene |
| UNG | UNG; HIGM4; EC 3.2.2; Uracil-DNA Glycosylase 2; Uracil-DNA Glycosylase; UNG15; UNG1; UDG; EC 3.2.2.27; Uracil-DNA Glycosylase 1, Uracil-DNA Glycosylase 2; UNG2; DGU; Uracil-DNA Glycosylase 1; HIGM5 | 7374 | P13051 | This gene encodes one of several uracil-DNA glycosylases. One important function of uracil-DNA glycosylases is to prevent mutagenesis by eliminating uracil from DNA molecules by cleaving the N-glycosylic bond and initiating the base-excision repair (BER) pathway. Uracil bases occur from cytosine deamination or misincorporation of dUMP residues. Alternative promoter usage and splicing of this gene leads to two different isoforms: the mitochondrial UNG1 and the nuclear UNG2. The UNG2 term was used as a previous symbol for the CCNO gene (GeneID 10309), which has been confused with this gene, in the literature and some databases. |
| XPA | XPA; XP1; Excision Repair-Controlling; XPAC; Mutant Xeroderma Pigmentosum Complementation Group A; Xeroderma Pigmentosum, Complementation Group A; Xeroderma Pigmentosum Group A-Complementing Protein; DNA Repair Protein Complementing XP-A Cells | 7507 | P23025 | Involved in DNA excision repair. Initiates repair by binding to damaged sites with various affinities, depending on the photoproduct and the transcriptional state of the region. Required for UV-induced CHEK1 phosphorylation and the recruitment of CEP164 to cyclobutane pyrimidine dimmers (CPD), sites of DNA damage after UV irradiation. |
| XRCC5 | XRCC5, KARP-1, KARP1, KU80, KUB2, Ku86, NFIV, Ku80, X-ray repair complementing defective repair in Chinese hamster cells 5, X-ray repair cross complementing 5 | 7520 | P13010 | The protein encoded by this gene is the 80-kilodalton subunit of the Ku heterodimer protein which is also known as ATP-dependant DNA helicase II or DNA repair protein XRCC5. Ku is the DNA-binding component of the DNA-dependent protein kinase, and it functions together with the DNA ligase IV-XRCC4 complex in the repair of DNA double-strand break by non-homologous end joining and the completion of V(D)J recombination events. This gene functionally complements Chinese hamster xrs-6, a mutant defective in DNA double-strand break repair and in ability to undergo V(D)J recombination. A rare microsatellite polymorphism in this gene is associated with cancer in patients of varying radiosensitivity. [provided by RefSeq, Jul 2008] |
| XRCC6 | XRCC6, X-ray repair complementing defective repair in Chinese hamster cells 6, CTC75, CTCBF, G22P1, KU70, ML8, TLAA, X-ray repair cross complementing 6 | 2547 | P12956 | The p70/p80 autoantigen is a nuclear complex consisting of two subunits with molecular masses of approximately 70 and 80 kDa. The complex functions as a single-stranded DNA-dependent ATP-dependent helicase. The complex may be involved in the repair of nonhomologous DNA ends such as that required for double-strand break repair, transposition, and V(D)J recombination. High levels of autoantibodies to p70 and p80 have been found in some patients with systemic lupus erythematosus. [provided by RefSeq, Jul 2008] |
Tested Data-Supported Products for Targeting DNA Repair
































Reference
- Helena, Jolene Michelle, et al. "Deoxyribonucleic acid damage and repair: Capitalizing on our understanding of the mechanisms of maintaining genomic integrity for therapeutic purposes." International journal of molecular sciences 19.4 (2018): 1148. Distributed under Open Access license CC BY 4.0, without modification.
For Research Use Only. Not For Clinical Use.