

Lorvotuzumab Mertansine Overview
Introduction of Lorvotuzumab Mertansine
Lorvotuzumab mertansine (IMGN901, BB10901, huN901-DM1) is an antibody-drug conjugate (ADC) composed of the cytotoxic maytansinoid derivative, DM1, conjugated to the humanized N901 monoclonal antibody (lorvotuzumab, huN901), which binds CD56 with high affinity. DM1 conjugated to lorvotuzumab via a stable disulfide SPP linker (When DM1 is attached to an antibody with the SPP linker, it is mertansine; when it is attached with the thioether linker, SMCC, it is emtansine). It was designed for the treatment of CD56 positive cancers (e.g. small-cell lung cancer, ovarian cancer). It has been granted orphan drug status for Merkel cell carcinoma and has reported encouraging Phase II results for small-cell lung cancer (SCLC). Once bound to CD56 on the surface of the target cell, the conjugate is internalized, the linker is cleaved, DM1 is released which in turn inhibits tubulin polymerization and results in cell death. Maytansine is a natural product, originally derived from the Ethiopian shrub Maytenus serrata. Maytansine inhibits tubulin polymerization, and is approximately 200-1,000 fold more cytotoxic than the Vinca alkaloids. Maytansine is clinically active but its narrow therapeutic window precluded further clinical development. Conjugation to an antibody and intracellular delivery could take advantage of the cytotoxic potency and expand the therapeutic window leading to greater tumor cell death with less overall toxicity. The maytansine derivative DM1 was developed specifically for use in ADCs.
Mechanism of Action of Lorvotuzumab Mertansine
CD56 is a membrane glycoprotein, also known as neural cell adhesion molecule (N-CAM), that is expressed on neural tissues and essentially all human natural killer (NK) cells and a subset of cytotoxic T cells. Its function has yet to be fully defined. CD56 is also expressed in various malignant tissues including small cell lung cancer, neuroblastoma, ovarian cancer and other neuroendocrine tumors. Interestingly, CD56 is not expressed in benign plasma cells and only mixed expression in the plasma cells of patients with monoclonal gammopathy of undetermined significance (MGUS). Conversely, CD56 is strongly expressed in up to 78% of multiple myeloma (MM). Although its function in MM cells is unknown, there are reports of CD56 positivity correlating with increased bone disease and loss of CD56 upon progression to plasma cell leukemia. DM1 is an antimitotic agent that inhibits microtubule assembly leading to cell cycle arrest and apoptosis. This agent is effective at picomolar concentrations. Although DM1 interact with the microtubules as the vinca alkaloid, it is 20-100 times more potent than vincristine. DM1 is also 24-270 times more potent than agents used in conventional chemotherapy, such as paclitaxel. The linker is a nonreducible thioether bond ([N-maleimidomethyl]cyclohexane-1-carboxylate) MCC that binds trastuzumab to DM1. The ideal linker should be stable in circulation to minimize the risk of systemic toxicities. However, the linker should efficiently release the cytotoxic agent upon ADC internalization. After binding to the CD56, the CD56 and drug-antibody conjugate are internalized via endocytosis. Intracellularly, DM1 is separated from the antibody by lysosomal degradation and binds to tubulin. This disrupts the microtubules causing cell cycle arrest and apoptosis. While lorvotuzumab is utilized to deliver DM1 to the cell, it also retains its properties of antibody-mediated cellular cytotoxicity (ADCC), and inhibition of CD56 signaling pathways.
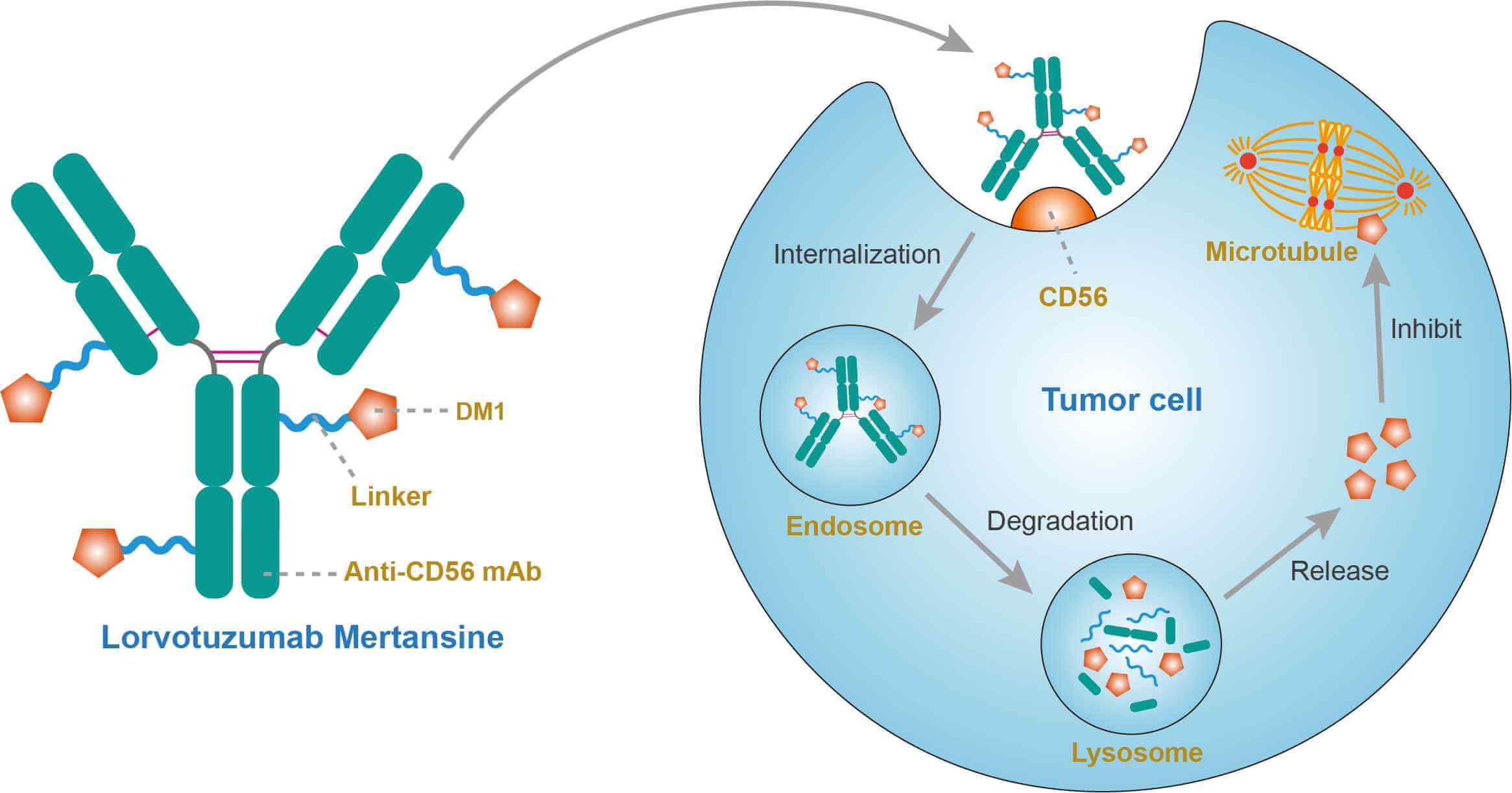 Fig.1 Mechanism of Action of Lorvotuzumab Mertansine
Fig.1 Mechanism of Action of Lorvotuzumab Mertansine
For research use only. Not intended for any clinical use.
Send Inquiry
This site is protected by reCAPTCHA and the Google Privacy Policy and Terms of Service apply.