

Flow Cytometry Protocol & Troubleshooting
Flow cytometry (FC) is a technique used to detect and analyze the chemical and physical properties of cells. In this procedure, labeled cells are suspended in a fluid and injected into a flow cytometer. Flow cytometry uses a laser as a light source to generate scattering and fluorescence signals.
FC is a powerful tool commonly used for various studies in the fields of molecular biology, microbiology bacteriology, and cancer biology. It can provide cell counting, sorting, functional analysis, microbial detection, etc. Creative Biolabs provides protocols for use in FC, including a step-by-step overview of the process. You can use this guide as an introductory guide or quick reference guide, and see our related products or services for more detailed information.
Solutions and Reagents
| Stages | Solutions and Reagents |
| Sample Preparation | Phosphate buffer (PBS), staining buffer, blocking buffer |
| Antibody Staining | Primary antibody, secondary antibody, antibody dilution buffer, fixative, permeabilizer, washing buffer |
Flow Cytometry Procedure
FC assays are generally based on 4 key steps. Paying attention to the small details will have a good impact on your result data.
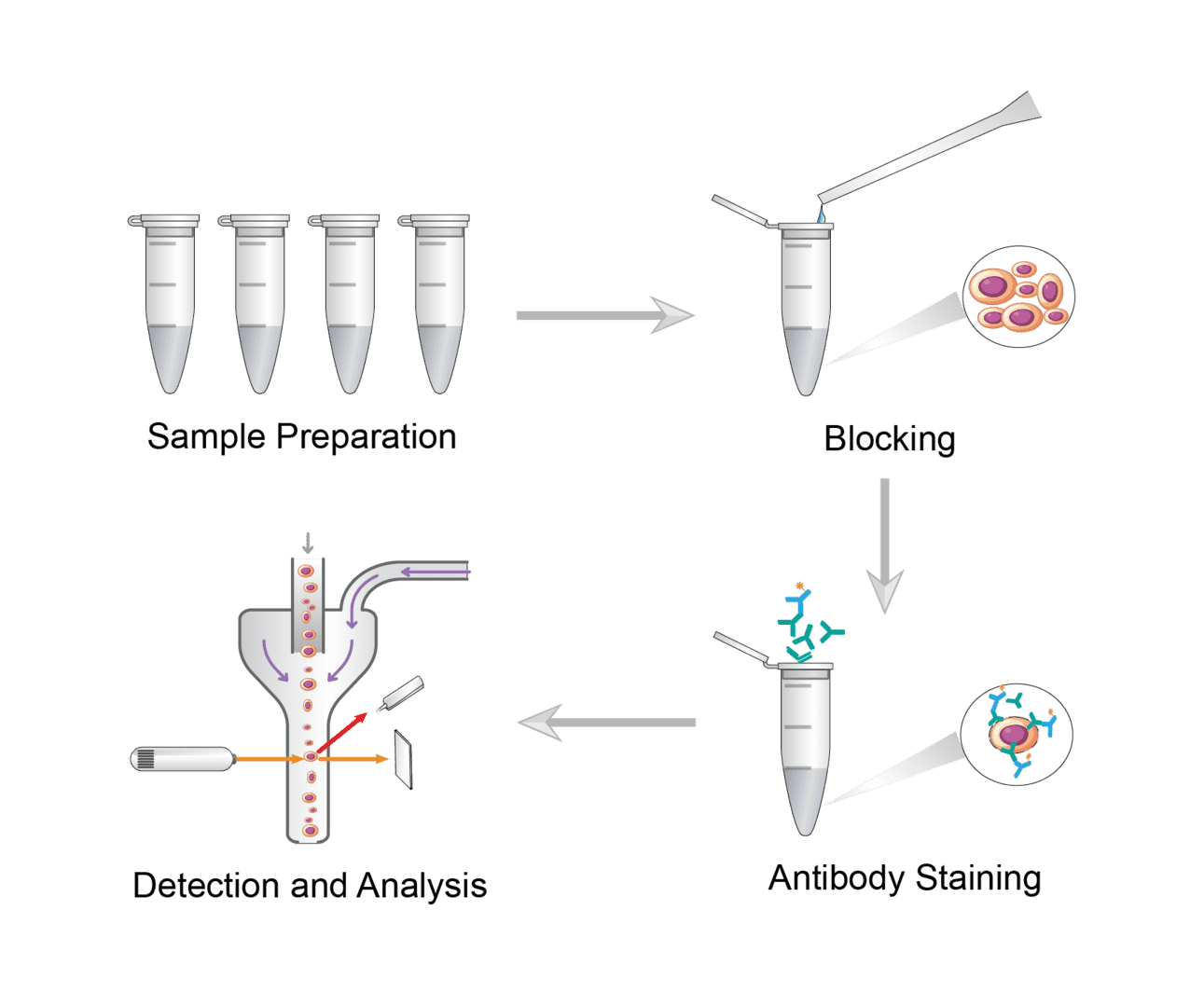
1. Sample Preparation
FC requires homogeneous single cell suspensions. Perform appropriate manipulations to obtain cell suspensions for adherent cells, non-adherent cells, tissue samples, etc. Mix the cell suspensions gently to ensure uniform mixing. Use a cell counter to obtain cell counts. Resuspend cells with staining buffer to the appropriate concentration.
2. Blocking
Blocking prevents non-specific binding of the antibody to the cell. Blocking agents can be selected based on the sample. Add a small amount of blocking agent to the cells for incubation. No washing step is required to ensure that blocking can be maintained throughout the process.
3. Antibody Staining
Perform immunofluorescence staining for cell surface antigens or intracellular antigens, respectively. For specific protocols, please refer to our other pages. We can use either direct staining or indirect staining. In direct staining, incubate the cells directly with fluorochrome-conjugated antibodies. In indirect staining, first incubate with the primary antibody and then incubate with the secondary antibody labeled by the fluorescent dye. The secondary antibody is specific for the primary antibody and can be detected. After staining, wash repeatedly to ensure no residual antibodies remain.
4. Detection and Analysis
Run samples on a flow cytometer and collect assay data. Analyze the data using flow cytometry data analysis software.
Troubleshooting
If your FC experiments are not producing quality results, check out our troubleshooting guide for tips on resolving common challenges.
Weak or no fluorescence signal
- Sample causes. First use freshly isolated cells whenever possible, rather than frozen samples. If using cryopreserved cells, check that the target antigen will not be affected by the freezing/thawing procedure. Next, check that the target protein is present in the cells and in sufficiently high amounts. For low expression antigens, use the brightest fluorescent dye or a two-step staining method to increase sensitivity. Finally, remember to perform all protocol steps at 4°C and use cold reagents.
- Fixation/permeabilization causes. If no signal is detected, it may be that your target antigen is inaccessible. Check that the fixation and permeabilization methods are correctly used for the target of interest. In addition to careful selection of the most appropriate fixative and permeabilization reagents, different procedures need to be used depending on the protein being detected.
- Antibody causes. If your signal is weak, your test antibody may be too dilute. We recommend increasing the amount or concentration of antibody. Or titrate the antibody before use to find the optimal amount for your particular experiment. You can also optimize antibody incubation time and temperature and consider using additional steps to amplify the signal. It is important to use secondary antibodies generated against the species from which the primary antibody was generated.
- Fluorescent dye causes. When selecting a fluorochrome, it is important to match the target used. It is important to control for any potential changes in fluorescence properties during the experiment, such as light, storage time, etc. Make sure that the fluorescence of the fluorescent dye is not fading at the time of the assay.
- Reagent causes. Take care to ensure that all reagents and solutions are of good quality and stored according to the manufacturer's instructions.
Excess fluorescent signal
- Antibody causes. High antibody concentrations can produce high non-specific binding or very high fluorescence intensity. You can reduce the amount of antibody added to each sample. It is better to titrate the antibody prior to use to find the optimal amount for a particular experiment.
- Fluorescent dye causes. Match the appropriate fluorescent dye to the antigen density.
- Blocking causes. Insufficient blocking causes excess signal due to non-specific binding. We recommend adding blocking agents and diluting antibodies in the blocking solution and increasing the blocking time.
- Washing causes. It may occur that unbound antibodies are trapped in the cells. Wash the cells well after each antibody incubation step.
High background
- Cell causes. One possibility is due to the presence of dead cells. A reactive dye can be used to exclude the dead cells. The other is due to high autofluorescence. Some cell types may naturally exhibit higher levels of autofluorescence. You can use fluorescent dyes that emit in the red-shift channel or very bright fluorescent dyes to solve the problem.
- Non-specific binding causes. The fact that the antibody targets a non-specific cell causes a high background. Block non-specific receptors on cells with blocking agents, BSA or FBS prior to antibody incubation.
- Antibody causes. Antibody overload causes high background. You can reduce the antibody concentration and use the recommended antibody dilutions to ensure the right dose of antibody. Optimize antibody concentration and incubation time based on cellular protein expression. If possible, avoid using biotinylated antibodies.
- Washing causes. Increase the number of washes after staining to ensure that excess antibody is removed.
Abnormal scatter profiles
- Cell causes. Samples should be fresh and properly prepared. Make sure that the cells in the sample are not lysed and broken. Do not centrifuge cells at high rotor speeds or vortex too vigorously. Perform a sieve on the cells to remove any dead cell debris. Also make sure that the sample is not contaminated with bacteria. Bacteria will automatically fluoresce at low levels. Finally, avoid storing stained cells for long periods of time.
The information in the above guide serves as a possible cause and solution to some of the most common problems in FC experiments. While the resources we have provided cover most of the problems encountered, this is not the only solution to some of the problems in your particular experiment. We are committed to helping our clients achieve better results and hope that this information will be useful.
If you need more help with your FC experiments, please contact us for assistance from our support team.
Products with Tested Data
At Creative Biolabs, we are dedicated to providing high-quality antibodies for various research applications. Each product in our extensive range has been rigorously tested to ensure superior reliability and efficacy. To showcase the performance of our antibodies, we have conducted numerous experiments using Flow Cytometry. Below, you will find a table listing a selection of our antibody products along with images from these experiments, demonstrating their proven reliability.
| Product Name | Catalog Number | Target | Image | Description |
|---|---|---|---|---|
| Human Anti-CLDN18 Recombinant Antibody (clone 25E3) | ZG-0140U | CLDN18 |

|
Untransfected KATO3 cells (green line), transfected Human CLDN18.2 KATO3 stable cells (red line) and transfected Human CLDN18.1 NIH-3T3 stable cells (blue line) were stained with anti-CLDN18 antibody (2µg/1*106 cells), washed and then followed by FITC-conjugated anti-Human IgG Fc antibody (ZG-0140U) and analyzed with flow cytometry. |
| Afuco™ Anti-CD3E ADCC Recombinant Antibody, ADCC Enhanced (AFC-TAB-H52) | AFC-TAB-H52 | CD3E |

|
Cell line transfected with CD3 was stained with Anti-CD3E Therapeutic Antibody (AFC-TAB-H52) NC1: FITC Isotype Ctrl FITC-CD3: FITC anti-human CD3 NC2: Human IgG1, lambda Isotype Control Antibody NC3: Blank AFC-TAB-H52: Afuco™ Anti-CD3E Therapeutic Antibody (Otelixizumab), ADCC Enhanced The secondary antibody: Goat Anti-human IgG FITC |
| Anti-Human CD20 Recombinant Antibody (TAB-028) | TAB-028 | MS4A1 |

|
Flow cytometry analysis of Daudi cells with Anti-CD20 Antibody (Cat#TAB-028, Blue line) or isotype control antibody (Yellow line). A Goat Anti-human IgG (FITC) was used as the secondary antibody. |
| Human Anti-CD22 Recombinant Antibody (TAB-1720CL) | TAB-1720CL | CD22 |

|
Flow cytometry analysis of Daudi cells with anti-CD22 antibody (Cat#TAB-1720CL, Blue line) or isotype control antibody (Yellow line). A Goat Anti-human IgG (FITC) was used as the secondary antibody. |
| Human Anti-CD22 Recombinant Antibody (TAB-1721CL) | TAB-1721CL | CD22 |

|
Flow cytometry analysis of Daudi cells with anti-CD22 antibody (Cat#TAB-1721CL, Blue line) or isotype control antibody (Yellow line). A Goat Anti-human IgG (FITC) was used as the secondary antibody. |
| Human Anti-CD22 Recombinant Antibody (TAB-1718CL) | TAB-1718CL | CD22 |

|
Flow cytometry analysis of Daudi cells with anti-CD22 antibody (Cat#TAB-1718CL, Blue line) or isotype control antibody (Yellow line). A Goat Anti-human IgG (FITC) was used as the secondary antibody. |
| Anti-Human CD22 Recombinant Antibody (RFB4) | TAB-1728CL | CD22 |

|
Flow cytometry analysis of Daudi cells with anti-CD22 antibody (Cat#TAB-1728CL, Blue line) or isotype control antibody (Yellow line). A Goat anti-Mouse IgG was used as the secondary antibody. |
| Humanized Anti-MS4A1 Recombinant Antibody (TAB-771) | TAB-771 | MS4A1 |

|
Flow cytometry analysis of Daudi cells with Anti-CD20 Antibody (Cat#TAB-771, Blue line) or isotype control antibody (Yellow line). A Goat Anti-human IgG (FITC) was used as the secondary antibody. |
| Human Anti-CD22 Recombinant Antibody (TAB-1717CL) | TAB-1717CL | CD22 |

|
Flow cytometry analysis of Daudi cells with anti-CD22 antibody (Cat#TAB-1717CL, Blue line) or isotype control antibody (Yellow line). A Goat Anti-human IgG (FITC) was used as the secondary antibody. |
| Human Anti-CD22 Recombinant Antibody (TAB-1722CL) | TAB-1722CL | CD22 |

|
Flow cytometry analysis of Daudi cells with anti-CD22 antibody (Cat#TAB-1722CL, Blue line) or isotype control antibody (Yellow line). A Goat Anti-human IgG (FITC) was used as the secondary antibody. |
| Anti-Human NOTCH3 Recombinant Antibody (TAB-562MZ) | TAB-562MZ | NOTCH3 |

|
Flow cytometry analysis of U87 MG cells with purified antibody TAB-562MZ (QCAb254, Blue). Cells were blocked with Human TruStain FcXTM and washed with 0.5% BSA-PBS. Cells were then incubated with TAB-562MZ (5 μl/5x10^5 cells) for 60 min at 4°C. The secondary antibody was FITC anti-human IgG antibody (3 μl) for 60 min at 4°C. |
| Recombinant Human Anti-CD20 Antibody (PABX-028) | PABX-028 | MS4A1 |

|
Flow cytometry analysis of Daudi cells with Anti-CD20 Antibody (Cat#PABX-028, Blue line) or isotype control antibody (Yellow line). A Goat Anti-human IgG (FITC) was used as the secondary antibody. |
| Human Anti-MS4A1 Recombinant Antibody (TAB-772) | TAB-772 | MS4A1 |

|
Flow cytometry analysis of Daudi cells with Anti-CD20 Antibody (Cat#TAB-772, Blue line) or isotype control antibody (Yellow line). A Goat Anti-human IgG (FITC) was used as the secondary antibody. |
| Anti-Human NOTCH3 Recombinant Antibody (TAB-559MZ) | TAB-559MZ | NOTCH3 |

|
Flow cytometry analysis of U87 MG cells with purified antibody TAB-559MZ (QCAb253, Blue). Cells were blocked with Human TruStain FcXTM and washed with 0.5% BSA-PBS. Cells were then incubated with TAB-559MZ (5 μl/5x10^5 cells) for 60 min at 4°C. The secondary antibody was FITC anti-human IgG antibody (3 μl) for 60 min at 4°C. |
| Human Anti-Human slc39a6 Antibody | MOB-143CQ | SLC39A6 |

|
Flow cytometry analysis of Jurkat cells with purified antibody MOB-143CQ (QCAb246, Blue). Cells were blocked with Human TruStain FcXTM and washed with 0.5% BSA-PBS. Cells were then incubated with MOB-143CQ (5 μl/5x10^5 cells) for 60 min at 4°C. The secondary antibody was PE anti-Human IgG antibody (3 μl) for 60 min at 4°C. |
| Anti-Human SLC1A5 Recombinant Antibody (TAB-1007CLV) | TAB-1007CLV | SLC1A5 |

|
Flow cytometry analysis of HT29 cells with purified antibody TAB-1007CLV (QCAb242, Blue). Cells were blocked with Human TruStain FcXTM and washed with 0.5% BSA-PBS. Cells were then incubated with TAB-1007CLV (5 μl/5x10^5 cells) for 60 min at 4°C. The secondary antibody was FITC anti-mouse IgG antibody (3 μl) for 60 min at 4°C. |
| Anti-Human NOTCH2 Recombinant Antibody (TAB-H67) | TAB-H67 | NOTCH2 |

|
Flow cytometry analysis of U87 MG cells with purified antibody TAB-H67 (QCAb249, Blue). Cells were blocked with Human TruStain FcXTM and washed with 0.5% BSA-PBS. Cells were then incubated with TAB-H67 (5 μl/5x10^5 cells) for 60 min at 4°C. The secondary antibody was FITC anti-human IgG antibody (3 μl) for 60 min at 4°C. |
| Human Anti-CD22 Recombinant Antibody (TAB-1719CL) | TAB-1719CL | CD22 |

|
Flow cytometry analysis of Daudi cells with anti-CD22 antibody (Cat#TAB-1719CL, Blue line) or isotype control antibody (Yellow line). A Goat Anti-human IgG (FITC) was used as the secondary antibody. |
| Anti-Human SLC1A5 Recombinant Antibody (TAB-1011CLV) | TAB-1011CLV | SLC1A5 |

|
Flow cytometry analysis of HT29 cells with purified antibody TAB-1011CLV (QCAb240, Blue). Cells were blocked with Human TruStain FcXTM and washed with 0.5% BSA-PBS. Cells were then incubated with TAB-1011CLV (5 μl/5x10^5 cells) for 60 min at 4°C. The secondary antibody was FITC anti-human IgG antibody (3 μl) for 60 min at 4°C. |
| Anti-Human SLC1A5 Recombinant Antibody (TAB-1010CLV) | TAB-1010CLV | SLC1A5 |

|
Flow cytometry analysis of HT29 cells with purified antibody TAB-1010CLV (QCAb241, Blue). Cells were blocked with Human TruStain FcXTM and washed with 0.5% BSA-PBS. Cells were then incubated with TAB-1010CLV (5 μl/5x10^5 cells) for 60 min at 4°C. The secondary antibody was FITC anti-human IgG antibody (3 μl) for 60 min at 4°C. |
| Mouse Anti-PECAM1 Recombinant Antibody (clone 7E8) | ZG-503C | PECAM1 |

|
FACS analysis of Thp-1 monocyte cell line with anti-CD31 clone 7E8. Control cells or cells stimulated with 10 ng/ml LPS were stained with anti-CD31 (1 μg/ml) in PBS+1% BSA and with goat anti-mouse-PE conjugate. The blue and red histograms show staining of cells without or with LPS, respectively. The grey histogram corresponds to the isotype control. |
| Anti-Human CD5 Recombinant Antibody (5D7) | TAB-308LC | CD5 |

|
Flow cytometry analysis of hPBMC cells incubated with Anti-Human CD5 Recombinant Antibody, Cat# TAB-308LC. hPBMC cells were stained with TAB-308LC (blue histogram, TE232276-1) or isotype control antibody (yellow histogram), followed by FITC-conjugated goat anti-Mouse IgG secondary antibody. The used concentration of TAB-308LC was 5 µg per 5 x 10^6 cells. |
| Human Anti-CD3 Recombinant Antibody scFv Fragment (HPAB-M0467-YC-S(P)) | HPAB-M0467-YC-S(P) | CD3 |
-4.jpg)
|
Flow cytometry analysis of Jurkat cells incubated with anti-human CD3 recombinant antibody cat# HPAB-M0467-YC-S(P). Jurkat cells were stained with HPAB-M0467-YC-S(P) (blue histogram, TE232382) or isotype control antibody (yellow histogram), followed by APC-conjugated goat anti-His tag secondary antibody. The used concentration of HPAB-M0467-YC-S(P) was 10 µg per 5 x 10^6 cells. |
| Recombinant Llama Anti-CD3 Single Domain Antibody (HPAB-M0378-YC) | HPAB-M0378-YC | CD3 |

|
Flow cytometry analysis of Jurkat cells incubated with anti-human CD3 recombinant antibody cat# HPAB-M0378-YC. Jurkat cells were stained with HPAB-M0378-YC (blue histogram, TE232382) or isotype control antibody (yellow histogram), followed by APC-conjugated goat anti-His tag secondary antibody. The used concentration of HPAB-M0378-YC was 10 µg per 5 x 10^6 cells. |
For research use only. Not intended for any clinical use.
Send Inquiry
This site is protected by reCAPTCHA and the Google Privacy Policy and Terms of Service apply.